Beactica is using its Eclipsor™ platform to build a pipeline of targeted protein degraders (TPDs) to address unmet medical needs in oncology.
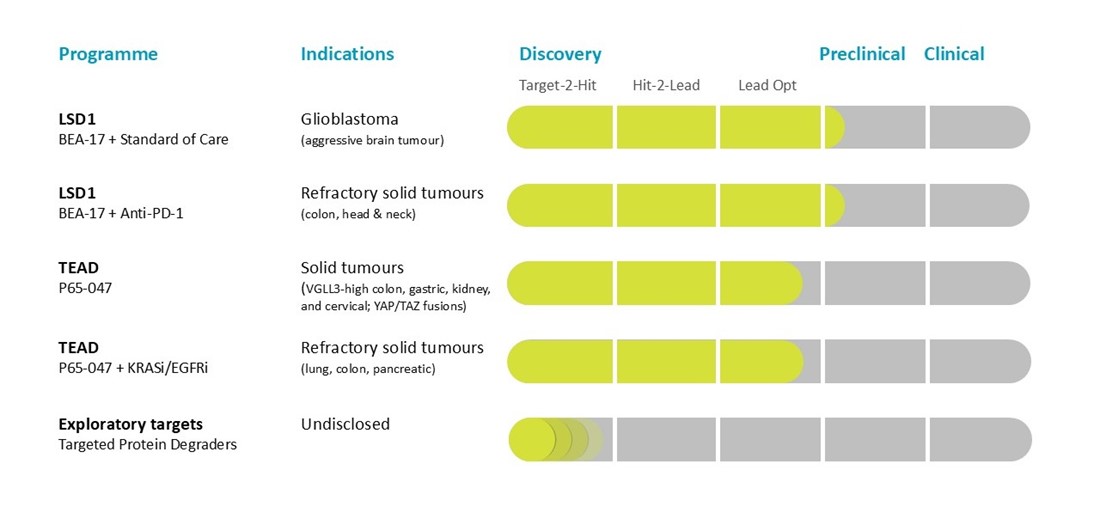
LSD1
Target Selection
Lysine-specific demethylase 1 (LSD1/KDM1A) is an epigenetic enzyme that regulates expression of large number of target genes and functions as a scaffolding protein that stabilizes transcriptional complexes. Furthermore, it is frequently overexpressed in cancer where it contributes to tumour growth. There is also strong evidence that inhibiting LSD1 function not only suppresses cancer growth, but also promotes anti-tumour immunity. The catalytic LSD1 inhibitors currently in clinical trials target the catalytic binding site on the protein and have a limited anti-cancer effect. Identifying alternative approaches that can also disrupt the scaffolding function of LSD1 is therefore of great importance.
Beactica’s approach
By leveraging a targeted approach, established technology and extensive expertise, we are developing a first-in-class small molecule scaffold inhibitor of the LSD1–CoREST complex. Unlike the catalytic LSD1 inhibitors that have shown limited anti-cancer effect in the clinic, our scaffold inhibitors exert their efficacy by inducing degradation of both LSD1 and its partner protein, CoREST. The lead compound, BEA-17, has been shown to potentiate the efficacy of checkpoint inhibitors in syngeneic models of colon cancer. It also potentiates glioblastoma standard-of-care (radiation + temozolomide) in syngeneic models of glioblastoma. Neither is observed with catalytic LSD1 inhibitors.
Improving lives
Beactica’s scaffold inhibitors of LSD1 have the potential to significantly increase the efficacy of immunomodulating cancer treatments such as checkpoint inhibitors and radiation.
The U.S. Food and Drug Administration (FDA) has granted Orphan Drug Designation to BEA-17 for the treatment of glioblastoma (GBM), the most common and aggressive form of brain tumour (read more here).
Selected posters & presentations
- Potentiation of Immunotherapy by LSD1 Modulation, American Association for Cancer Research (AACR) Annual Meeting 2023, Orlando, Florida. Sunday, 16 April, 2023.
Related press releases
- Beactica and KU Leuven secure EUR 2.5 million from the European Innovation Council to advance orphan drug BEA-17 to clinical readiness
- Beactica Therapeutics receives FDA Orphan Drug Designation for BEA-17 for the treatment of glioblastoma
Key collaborators

Further reading
- Sheng W et al. (2018) LSD1 Ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell, 174:1–15.
- Qin Y et al. (2018) Inhibition of histone lysine-specific demethylase 1 elicits breast tumor immunity and enhances antitumor efficacy of immune checkpoint blockade. Oncogene, 38:390–405.
- Sehrawat A et al. (2018) LSD1 activates a lethal prostate cancer gene network independently of its demethylase function. PNAS, 115:E4179–E4188.
YAP–TEAD
Target Selection
YAP1 (Yes-associated protein 1) is a coactivator that together with TEAD (TEA Domain) transcription factors play key roles in the Hippo signalling pathway that regulate cell proliferation, apoptosis, and stemness. Dysregulation of the Hippo pathway and subsequent activation of TEAD has been reported in a wide range of cancers such as squamous cell carcinoma, head and neck, gynaecological, and gastrointestinal cancers. TEAD has four paralogs, at least one of which is synthetically lethal (TEAD1) in certain genetic settings. The predominate approach pursued by pharmaceutical companies to silence TEAD is to bind to the lipid pocket thereby preventing the S-palmitoylation required for YAP activation of TEAD.
Beactica’s approach
We are utilising our Eclipsor™ platform for targeted protein degradation to develop novel therapeutics for treatment of cancers caused by dysregulation of the Hippo pathway. Lead compound P65-047 is a first-in-class proteolysis-targeting degrader of TEAD transcription factors that outperforms competing palmitoylation inhibitors currently in clinical development.
Improving lives
P65-047 is being developed as stand-alone product in niche cancers and combination therapy for underserved subgroups in common solid tumours.
Selected posters & presentations
- P65-047, a novel TEAD degrader, overcomes KRAS inhibitor resistance through Hippo pathway disruption in NSCLC, American Association for Cancer Research (AACR) Annual Meeting 2026, San Diego, California. Tuesday, 22 April, 2026.
- Expanding the TEAD Therapeutic Potential with Degraders: In Vitro Sensitivity, Predictive Biomarkers, and In Vivo Efficacy, American Association for Cancer Research (AACR) Annual Meeting 2025, Chicago, IL. Sunday, 28 April, 2025.
- Degraders of TEAD Transcription Factors Based on Interface 3 Binders, American Association for Cancer Research (AACR) Annual Meeting 2024, San Diego, California. Sunday, 7 April, 2024.
Related press releases
- Beactica Therapeutics announces presentation at the American Association for Cancer Research Annual Meeting 2026 on its TEAD degrader programme
- Beactica Therapeutics announces collaboration with the National Center for Advancing Translational Sciences at NIH
Key collaborators
 |  |  |
Further reading
- Calses PC et al. (2019) Hippo pathway in cancer: Aberrant regulation and therapeutic opportunities. Trends in Cancer, 5:297–307.
- Cunningham R et al. (2022) The Hippo pathway in cancer: YAP/TAZ and TEAD as therapeutic targets in cancer. Clin Sci (Lond), 136:197–222.
- Pobbati AV et al. (2020) Protein-protein interaction disruptors of the YAP/TAZ-TEAD transcriptional complex. Molecules, 25:6001–6017.
- Furet P et al. (2022) The first class of small molecules potently disrupting the YAP-TEAD interaction by direct competition. ChemMedChem, e202200303.
Exploratory Targets
Beactica is continually evaluating new disease-causing target proteins with therapeutic breakthrough potential to add to its pipeline.
Targeted Protein Degradation
Beactica is leveraging targeted protein degradation to selectively eliminate disease-causing proteins.
Stay Updated
Sign up for the Beactica newsletter to receive our latest news and updates